Alzheimer’s Disease and Synapse Biology (2016–Present)
work in progress
Early Mechanobiology and Neuroengineering Works ('03–'15)
Mechanochemical control of axonal growth cones
Background: The glial scar that forms following spinal cord injury is a source of axon repellents. Axon towing, modulating axon growth by directly pulling on the growth cones, has been demonstrated previously. We designed an in vitro experiment to test axons could be towed with physiologically relevant forces (< 10 pN) towards a source of axon repellents.
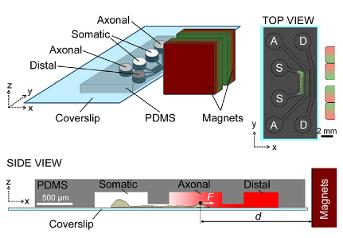
Methods: Primary mouse cortical neurons were cultured in tricompartmental microfluidic devices. A linear concentration gradient forms if a compound is only added to the distal compartment. Magnetic tweezers forces were applied on growth cones that were targeted by magnetic beads coated with an antibody against NCAM. Axons were treated locally with a panel of inhibitors.
Major Findings:
- Axons subjected to Semaphorin 3A (Sema3A) and chondroitin sulfate proteoglycans (CSPG) gradients slow down and deviate from their elongation direction.
- Inhibition of axonal Rho kinase (ROCK) and calpain blocks the effects of Sema3A. Inhibition of ROCK and kinesin-5 blocks the effects of CSPG.
- Growth cone pulling with less than 12 pN force acting on NCAMs successfully towed axons against Sema3A gradients only when axons were co-treated with ROCK inhibitors.
- Similarly, axons were towed against CSPG when they were co-treated with inhibitors of ROCK and kinesin-5.
Significance: This study shows that CNS axons can be towed with much less force than previously thought. This suggest that axon towing may be developed towards a minimally-invasive therapy for rewiring the CNS following traumatic injury. This cartoon summarizes the possible underlying mechanisms of mechano-chemical stimulation of axonal growth cones.
Cellular mechanotransduction with FeAu nanorods
Background: Targeting of cells based on their receptor expression may enable selective treatment modalities. FeAu nanorods may be exploited for their optical and magnetic properties.
Methods: Fe segments were coated with PEG. Au tips were functionalized with Streptavidin (STR) or with Heregulin (HRG). Cell targeting was tested under flow conditions in a microfluidic channel. Magnetic tweezers forces were applied and ERK phosphorylation was assessed using immunocytochemistry.
Major Findings:
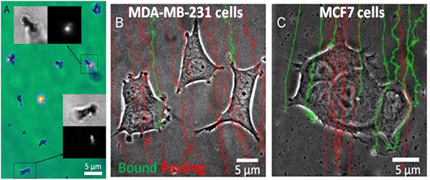
- FITC-biotin labeling of STR-conjugated nanorods confirms tip functionalization (A).
- MCF7 cells express ErbB2 receptors 2.3× higher in MCF7 cells. Accordingly, HRG-rods binding to MCF7 is 2.2× higher than MDA cells (B-C).
- MCF7 cells targeted with HRG-nanorods exhibit focal ERK phosphorylation. These ‘active zones’ were also targeted preferentially, 6× more often than the rest of the cell.
- Periodic mechanical stretch (0.5 Hz) enhances ERK phosphorylation in targeted cells regardless of the force magnitude (4.1-12.7 pN). At high forces, rods formed membrane tethers.
- Separately, low dose B-Raf inhibition enhances ERK activation and kills MCF7 cells when combined with periodic mechanical stretch.
- Magnetic hyperthermia and subsequent cell death can be induced with very weak alternating magnetic fields and at low particle densities.
Significance: FeAu nanorods are excellent tools for cell targeting and force application in vitro. Combining mechanical and pharmaceutical treatement induces ERK hyperstimulation and subsequent cell death.
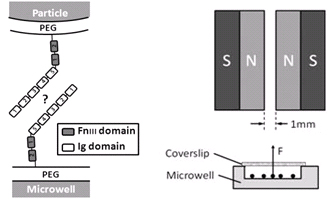
apCAM bond mechanics using magnetic tweezers
Background: Aplysia cell adhesion molecule (apCAM) is a structural and functional homolog of human neural CAM. Its homophilic bond is crucial for synapse formation and maturation. The role of apCAM in growth cone mechano-transduction is well established, the mechanics of apCAM bond has yet to be characterized. The extracellular portion of apCAM consist of two fibronectin (Fn) and five immunoglobulin-like (Ig) domains. Their roles in bond formation are not clear.
Methods: We functionalized magnetic particles and flat surfaces with oriented apCAM via NTA/His6 chemistry. We applied constant vertical forces to the particles and measured bond lifetimes.
Major Findings:
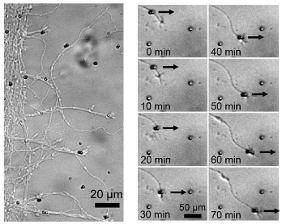
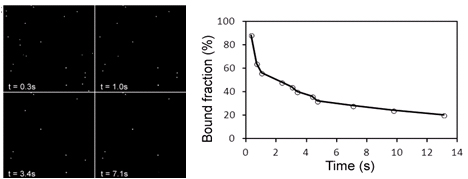
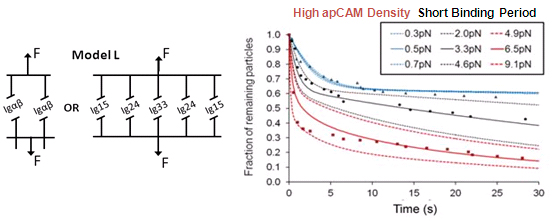
- External force decreases bond lifetime as a function of force magnitude, surface density, and binding period.
- Simulations based on Bell model, koff = koff,0 · exp(Fext/f), favor a bond model with two types of bonds.
Significance: This study was the first to pull on Ig-CAM bonds with constant, physiologically relevant forces. Bond dissociation rate increased according to Bell model. Magnetic tweezers–based force clamp was shown to be suitable method to characterize weak molecular interactions.
Massively parallel axotomy using microfluidics
Background: Wallerian degeneration (WD) is a series of events that occur in distal axons when they are axotomized. At late stages, WD shares similarities with other neurodegenerative diseases and therefore is a relevant experimental paradigm to study axonal degeneration mechanisms. WD is an active, caspase-independent self-destruction process where a wave of axonal fragmentation propagates rapidly along individual axons. WD proceeds anterograde and axons degenerate asynchronously.
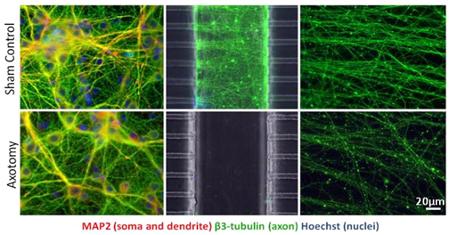
Methods: We developed a microfluidic culture device that enables localized central axotomy in numerous CNS neurons simultaneously. It provides exclusive access to somatic and axonal chambers independently before and after the axotomy.
Major Findings:
- Axotomy induces progressive degeneration in distal axons, but no cell death in 24h. Degeneration is asynchronous and progresses anterograde.
- Axotomized axons were treated with post-injury calpain and caspase inhibitors and pre- and post-injury βNAD. Wallerian-like degeneration is delayed with βNAD but not with calpain nor caspase inhibitors.
Significance: Our model enables simultaneous mass axotomy of CNS axons and permits the treatment of isolated segments along the axons. Wallerian-like degeneration can be blocked by pre-treating with βNAD, a molecule associated with mitochondrial energy metabolism and neurodegenerative diseases.
Cellular mechanisms of traumatic axonal injury
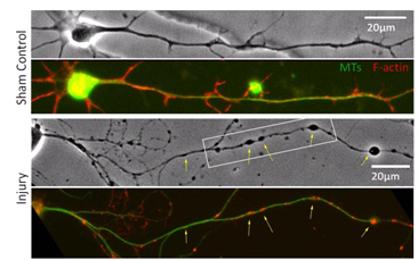
Background: Human diffuse axonal injury (DAI) is a result of closed head trauma and exhibits (i) increased membrane permeability, (ii) increased [Ca2+] and calpain activity, (iii) cytoskeletal damage and impaired axonal transport, and (iv) axonal beading, the “hallmark” morphology of DAI.
Methods: We applied fluid shear stress injury to cultured central nervous system (CNS) neurons in order to mimic axon degeneration following DAI. To test the hypothesis, we
- treated injured neurons with Poloxamer 188 (P188), a triblock copolymer that reseals damaged membranes
- chelated intra- and extra-cellular Ca2+ using BAPTA and EGTA, respectively
- inhibited calpain activity using its specific inhibitor ALLN
Major Findings:
- Injury induces membrane poration and gradual axonal beading. Post-injury P188 application blocks both responses. Focal microtubule disruption underlies bead formation.
- Injury induces increases in [Ca2+]i and calpain which are blocked by Ca2+ chelation, calpain inhibition, and by post-injury P188 treatment.
- Axonal mitochondria (“hot spots” in CellTracker image of uninjured axon) predict future bead locations.
Significance: Our model mimics important features of DAI including mechanoporation, microtubule disruption, and axonal beading. We provide evidence that Ca2+-mediated calpain activity is a crucial step in the DAI neuropathology. The co-localization of the Ca2+ hot spots, mitochondria, and future bead locations suggests that mitochondria play an important role in the focal cytoskeletal damage following injury. Finally, we propose P188 as a membrane therapy for traumatic injury.